Enzyme Replacement Therapy
In 2006 the commercial availability of ERT made Pompe disease the first inheritable muscle disorder for which therapy is available (van den Hout et al. 2000; Kishnani et al. 2007; van der Ploeg et al. 2010). For a few other lysosomal storage disorders (Gaucher disease, Fabry disease, mucopolysaccharidosis I, II, and VI), similar therapies became available at earlier stages (Barton et al. 1991; Eng et al. 2001; Schiffmann et al. 2001; Kakkis et al. 2001; Harmatz et al. 2004).
The only treatment available currently, for Pompe Disease patients, is a process called Enzyme Replacement Therapy (ERT), where they are administered with an enzyme called Myozyme (alglucosidase alfa). It has been shown, in clinical trials with infantile-onset patients, to decrease heart size, maintain normal heart function, improve muscle function, tone, and strength, and reduce glycogen accumulation (Klinge, Straub & Neudorf, 2005). Myozyme is administered by intravenous (IV) infusion. This is a process that involves injecting the drug into a vein, directly into the bloodstream. The total volume of infusion is determined by the patient’s body weight and should be administered over approximately 4 hours. Myozyme should be administered by a healthcare professional and patients must be monitored during the course and even the 2 hours that follow, in case of allergies or any other possible reactions to the treatment.
 A lifeline nevertheless, Enzyme Replacement Therapy (ERT) extends survival and improves the quality of life in Pompe disease. However, costs of ERT in Pompe disease are considerably high, especially since the dosage required by an individual patient is determined by his/her body weight. Traditional cost-effectiveness analysis is considered inappropriate for orphan diseases because of their low frequencies and relatively high developmental costs of treatment (Drummond et al. 2007). Cost-effectiveness is, however, a topic that should be addressed given the conditions of reimbursement of ERT. Most cost of illness studies often performed from a societal perspective, fail to account for the entire burden of an illness. Patients might need medical devices; they might need home care and informal care (which can potentially also affect patients’ families’ lives); patients’ capacity to work could be limited, and any disease can affect patients’ health-related quality of life (HRQoL). A burden of illness (BOI) study, which compiles all these aspects, has been unavailable for adult Pompe disease patients, although the BOI of adults with Pompe disease is expected to be substantial. Hence, the aim of this study is to assess the burden of Pompe disease with respect to all aspects: societal costs, the use of home care and informal care, productivity losses, and losses in HRQoL. Following Hagemans et al. (2004), quality of life is expected to be lower for more severely affected patients, i.e., patients using ambulatory or respiratory devices. These patients are also expected to incur higher annual costs.
A lifeline nevertheless, Enzyme Replacement Therapy (ERT) extends survival and improves the quality of life in Pompe disease. However, costs of ERT in Pompe disease are considerably high, especially since the dosage required by an individual patient is determined by his/her body weight. Traditional cost-effectiveness analysis is considered inappropriate for orphan diseases because of their low frequencies and relatively high developmental costs of treatment (Drummond et al. 2007). Cost-effectiveness is, however, a topic that should be addressed given the conditions of reimbursement of ERT. Most cost of illness studies often performed from a societal perspective, fail to account for the entire burden of an illness. Patients might need medical devices; they might need home care and informal care (which can potentially also affect patients’ families’ lives); patients’ capacity to work could be limited, and any disease can affect patients’ health-related quality of life (HRQoL). A burden of illness (BOI) study, which compiles all these aspects, has been unavailable for adult Pompe disease patients, although the BOI of adults with Pompe disease is expected to be substantial. Hence, the aim of this study is to assess the burden of Pompe disease with respect to all aspects: societal costs, the use of home care and informal care, productivity losses, and losses in HRQoL. Following Hagemans et al. (2004), quality of life is expected to be lower for more severely affected patients, i.e., patients using ambulatory or respiratory devices. These patients are also expected to incur higher annual costs.
Supportive therapies
Most patients require supportive therapy to address the symptoms of Pompe disease, which include respiratory and cardiac problems, physical disability, and difficulty in swallowing. Patients, especially those diagnosed late with weakening skeletal muscles, usually require a mechanical ventilator during respiratory infections or during sleep.
Patients with physical weakness will need physical and occupational therapy can improve their muscle strength and help them to learn to use canes or walkers if necessary. Speech therapy may be helpful if a patient’s mouth and tongue muscles are affected. Weakness in these muscles can also make it difficult to swallow, leading to problems in getting adequate nutrition.
Infant onset Pompe disease patients may need to use a feeding tube or feeding button. Patients with late-onset Pompe typically do not need such assistance but they may benefit from a soft diet.
Future therapies
The future is looking bright for Pompe disease patients as there are several variations of ERT currently undergoing clinical trials to become the next Pompe disease treatments. Currently, researchers are exploring the possibility of medications that act as “chaperones” to the 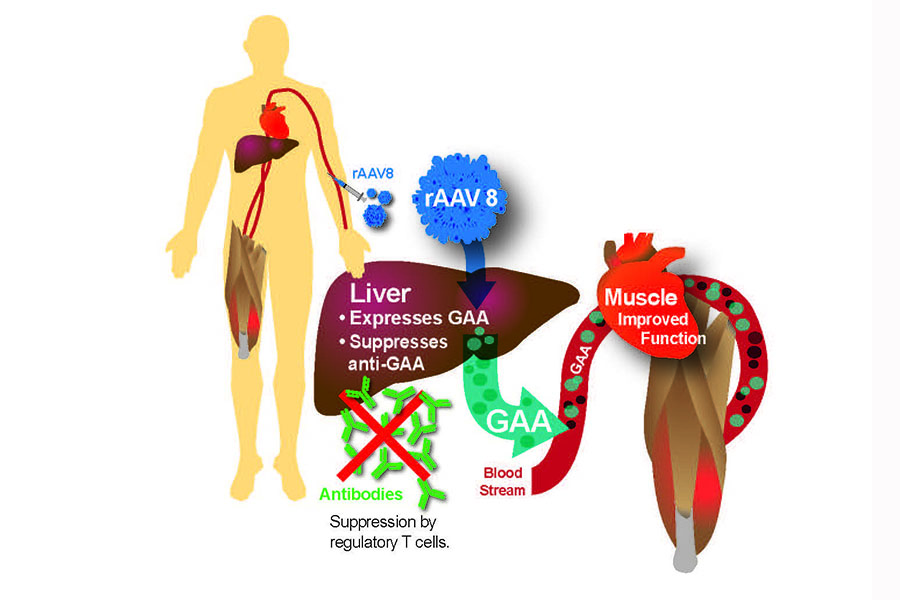 patient’s available GAA enzyme, so as to make the enzyme more effective at breaking down glycogen.
patient’s available GAA enzyme, so as to make the enzyme more effective at breaking down glycogen.
Researchers at Duke Medical are working on a gene therapy approach that could allow Pompe disease patients to produce their own functional GAA enzyme, a step that would help avoid immune reactions to ERT, and aid patients for whom ERT is not effective.